

mRNA-Sequenzierung in voller Länge – PacBio





Servicevorteile
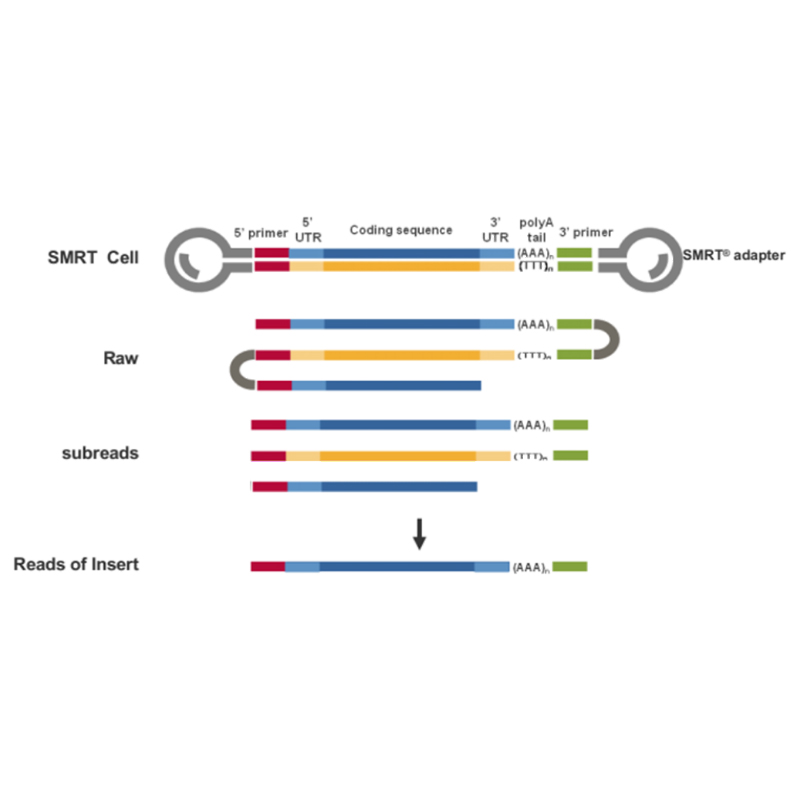
● Direktes Auslesen des cDNA-Moleküls voller Länge vom 3'-Ende zum 5'-Ende
● Auflösung auf Isoformebene in der Sequenzstruktur
● Transkripte mit hoher Genauigkeit und Integrität
● Hohe Verträglichkeit mit verschiedenen Arten
● Große Sequenzierungskapazität mit 4 ausgestatteten PacBio Sequel II-Sequenzierungsplattformen
● Sehr erfahren mit über 700 Pacbio-basierten RNA-Sequenzierungsprojekten
● BMKCloud-basierte Ergebnisbereitstellung: Maßgeschneidertes Data-Mining auf der Plattform verfügbar.
● Kundendienstleistungen gültig für 3 Monate nach Projektabschluss
Leistungsbeschreibung
Plattform: PacBio Sequel II
Sequenzierungsbibliothek: Poly A-angereicherte mRNA-Bibliothek
Empfohlener Datenertrag: 20 GB/Probe (abhängig von der Art)
FLNC(%): ≥75%
*FLNC: Nicht-chimäre Transkripte in voller Länge
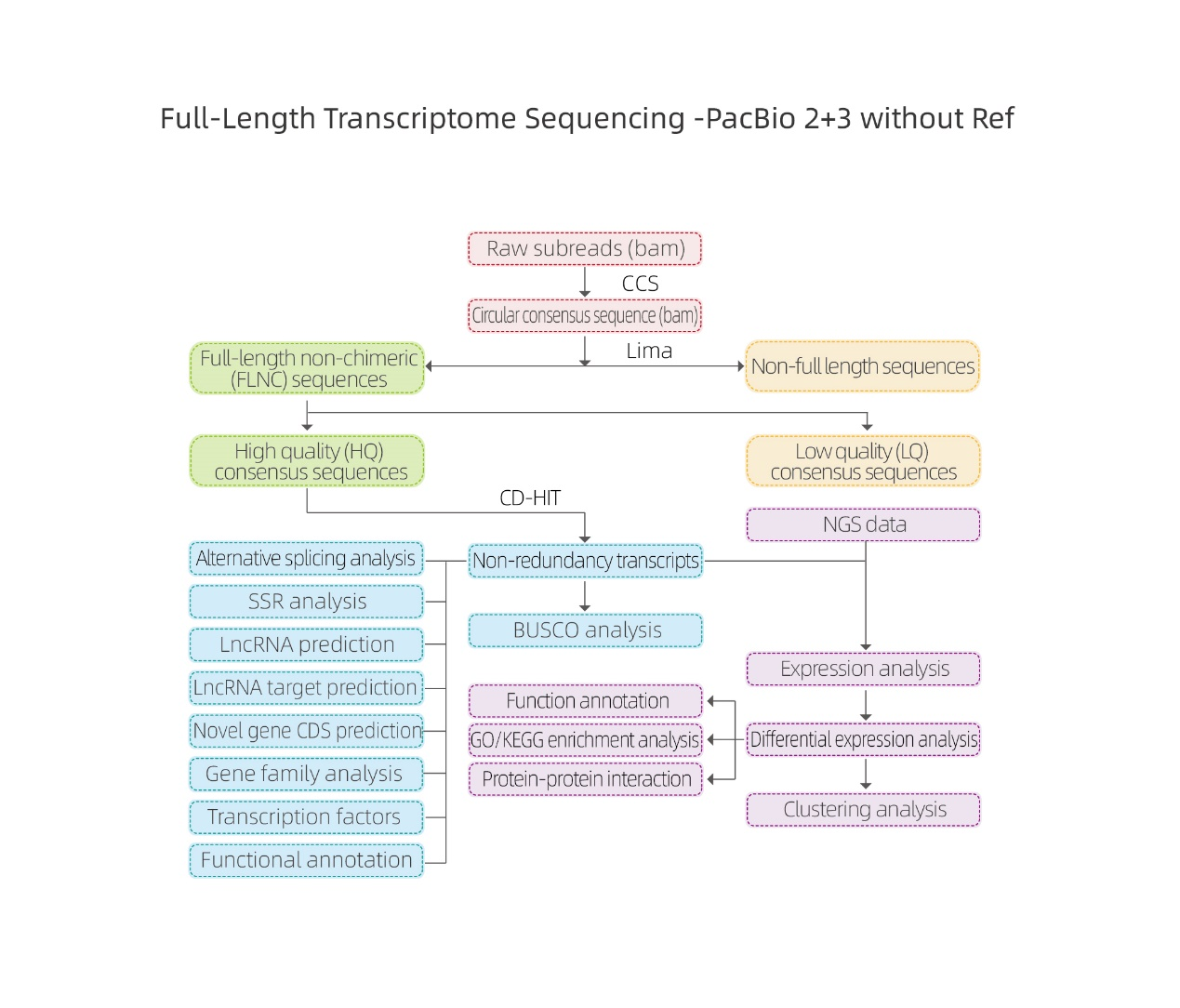
Bioinformatische Analysen
● Rohdatenverarbeitung
● Transkriptidentifizierung
● Sequenzstruktur
● Expressionsquantifizierung
● Funktionsanmerkung
Musteranforderungen und Lieferung
Probenanforderungen:
Nukleotide:
| Konz. (ng/μl) | Menge (μg) | Reinheit | Integrität |
| ≥ 120 | ≥ 0,6 | OD260/280=1,7-2,5 OD260/230=0,5-2,5 Begrenzte oder keine Protein- oder DNA-Kontamination auf dem Gel sichtbar. | Für Pflanzen: RIN≥7,5; Für Tiere: RIN≥8,0; 5,0≥ 28S/18S≥1,0; begrenzte oder keine Grundlinienhöhe |
Gewebe: Gewicht (trocken):≥1 g
*Für Gewebe mit weniger als 5 mg empfehlen wir, schockgefrorene (in flüssigem Stickstoff) Gewebeproben einzusenden.
Zellsuspension:Zellzahl = 3×106- 1×107
*Wir empfehlen den Versand von gefrorenem Zelllysat.Für den Fall, dass die Zellenzahl kleiner als 5×10 ist5Es wird empfohlen, in flüssigem Stickstoff schockgefrostet zu werden, was für die Mikroextraktion vorzuziehen ist.
Blutproben:Volumen ≥ 1 ml
Mikroorganismus:Masse ≥ 1 g
Empfohlene Musterlieferung
Container:
2 ml Zentrifugenröhrchen (Alufolie wird nicht empfohlen)
Probenbeschriftung: Gruppe+Replikation, z. B. A1, A2, A3;B1, B2, B3... ...
Sendung:
1. Trockeneis: Proben müssen in Beutel verpackt und in Trockeneis vergraben werden.
2. RNAstable-Röhrchen: RNA-Proben können in RNA-Stabilisierungsröhrchen (z. B. RNAstable®) getrocknet und bei Raumtemperatur versendet werden.
Service-Workflow

Experimentdesign

Musterlieferung

RNA-Extraktion

Bibliotheksbau

Sequenzierung

Datenanalyse

Kundendienst
1.FLNC-Längenverteilung
Die Länge des nicht-chimären Lesevorgangs in voller Länge (FLNC) gibt die Länge der cDNA in der Bibliothekskonstruktion an.Die FLNC-Längenverteilung ist ein entscheidender Indikator für die Bewertung der Qualität des Bibliotheksaufbaus.
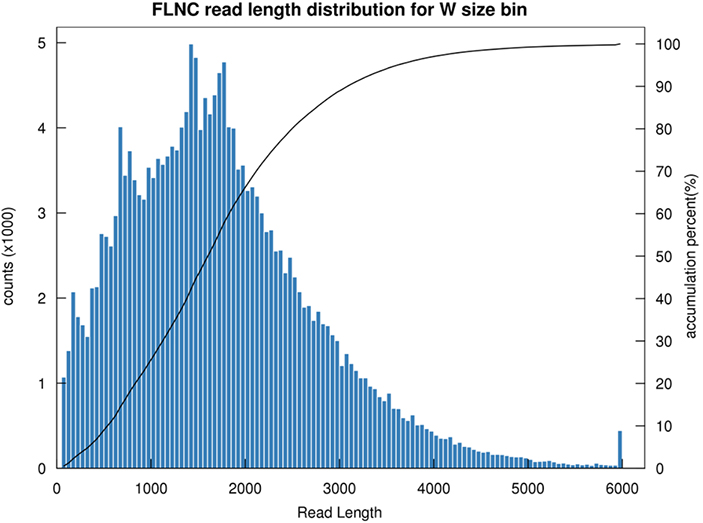
FLNC-Leselängenverteilung
2. Vollständige Längenverteilung der ORF-Region
Wir verwenden TransDecoder, um proteinkodierende Regionen und entsprechende Aminosäuresequenzen vorherzusagen und so Unigene-Sets zu generieren, die in allen Proben vollständige, nicht-redundante Transkriptinformationen enthalten.
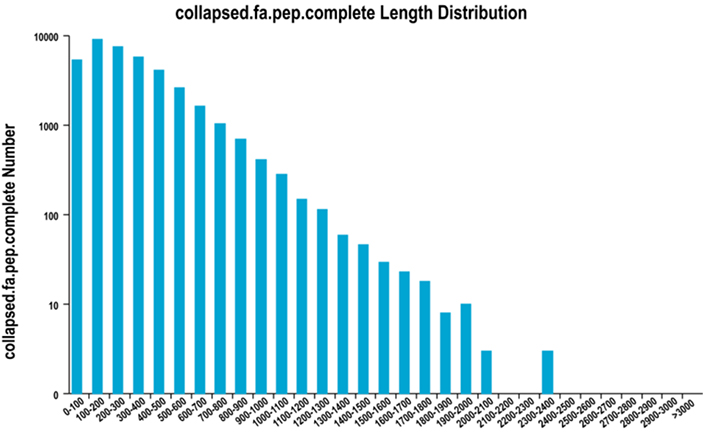
Vollständige Längenverteilung der ORF-Region
3. Analyse der Anreicherung des KEGG-Signalwegs
Differenziell exprimierte Transkripte (DETs) können identifiziert werden, indem NGS-basierte RNA-Sequenzierungsdaten mit Transkriptsätzen voller Länge abgeglichen werden, die durch PacBio-Sequenzierungsdaten generiert wurden.Diese DETs können für verschiedene Funktionsanalysen weiterverarbeitet werden, z. B. für die KEGG-Signalweganreicherungsanalyse.
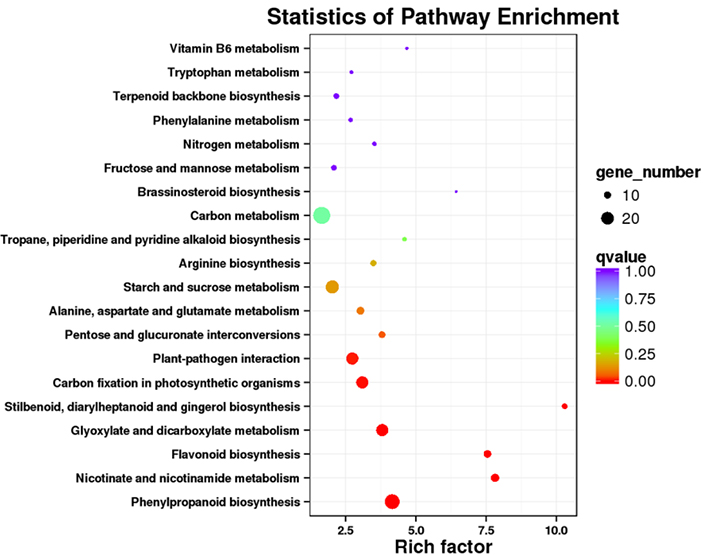
Anreicherung des DET KEGG-Signalwegs – Punktdiagramm
BMK-Fall
Die Entwicklungsdynamik des Populus-Stammtranskriptoms
Veröffentlicht: Zeitschrift für Pflanzenbiotechnologie, 2019
Sequenzierungsstrategie:
Beispielsammlung:Stammregionen: Spitze, erstes Internodium (IN1), zweites Internodium (IN2), drittes Internodium (IN3), Internodium (IN4) und Internodium (IN5) von Nanlin895
NGS-Sequenz:Die RNA von 15 Personen wurde als eine biologische Probe zusammengefasst.Drei biologische Replikate jedes Punktes wurden für die NGS-Sequenz verarbeitet
TGS-Sequenz:Die Stammregionen wurden in drei Regionen unterteilt, nämlich Apex, IN1-IN3 und IN4-IN5.Jede Region wurde für die PacBio-Sequenzierung mit vier Arten von Bibliotheken verarbeitet: 0–1 kb, 1–2 kb, 2–3 kb und 3–10 kb.
Wichtigste Ergebnisse
1. Insgesamt wurden 87.150 Transkripte voller Länge identifiziert, in denen 2.081 neue Isoformen und 62.058 neue alternative gespleißte Isoformen identifiziert wurden.
Es wurden 2.1187 lncRNA- und 356 Fusionsgene identifiziert.
3. Vom Primärwachstum bis zum Sekundärwachstum wurden 15838 unterschiedlich exprimierte Transkripte von 995 unterschiedlich exprimierten Genen identifiziert.In allen DEGs waren 1216 Transkriptionsfaktoren, von denen die meisten noch nicht gemeldet wurden.
Die 4.GO-Anreicherungsanalyse zeigte die Bedeutung der Zellteilung und des Oxidations-Reduktionsprozesses für das Primär- und Sekundärwachstum.
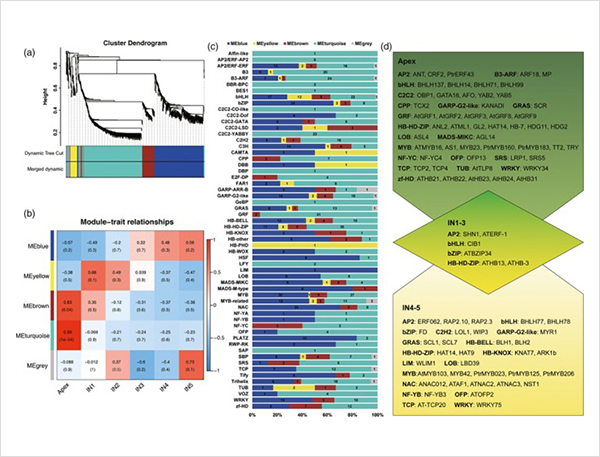
Alternative Spleißereignisse und verschiedene Isoformen
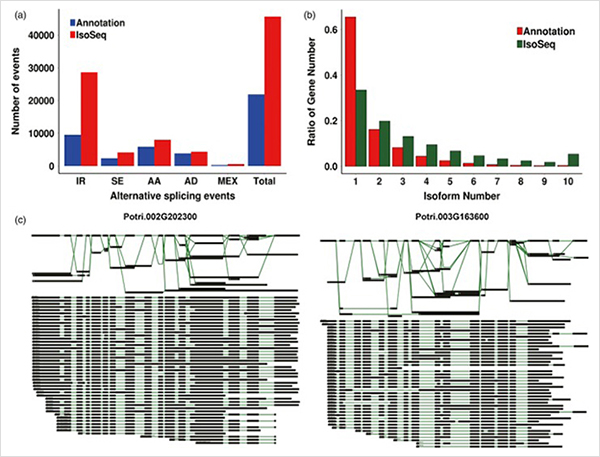
WGCNA-Analyse zu Transkriptionsfaktoren
Referenz
Chao Q, Gao ZF, Zhang D, et al.Die Entwicklungsdynamik des Populus-Stammtranskriptoms.Plant Biotechnol J. 2019;17(1):206-219.doi:10.1111/pbi.12958





