

Vysoce výkonné genotypování, zejména na rozsáhlé populaci, je základním krokem v genetických asociačních studiích, které poskytují genetický základ pro objev funkčních genů, evoluční analýzu atd. Namísto hlubokého resekvenování celého genomu, redukované sekvenování genomu (RRGS ) je zaveden s cílem minimalizovat náklady na sekvenování na vzorek a zároveň zachovat rozumnou účinnost při objevování genetických markerů.Toho se běžně dosahuje extrakcí restrikčních fragmentů v daném rozsahu velikostí, který se nazývá knihovna redukované reprezentace (RRL).Specific-locus amplified fragment sekvencování (SLAF-Seq) je samostatně vyvinutá strategie pro de novo objev SNP a SNP genotypování velkých populací.
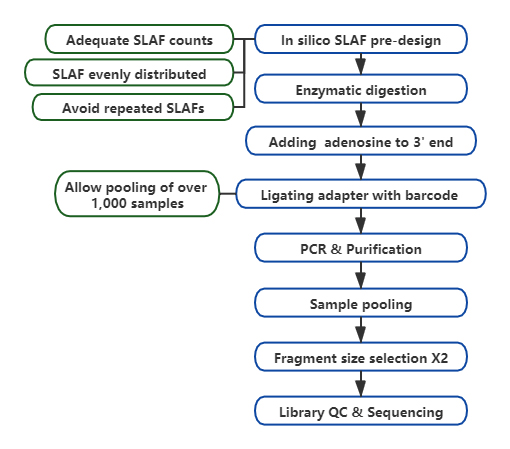
Technický pracovní postup
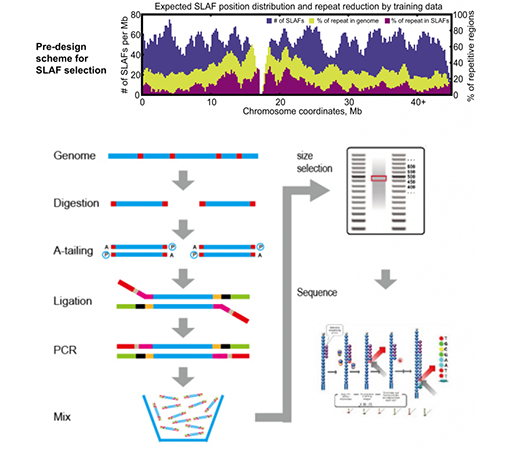
SLAF vs stávající metody RRL
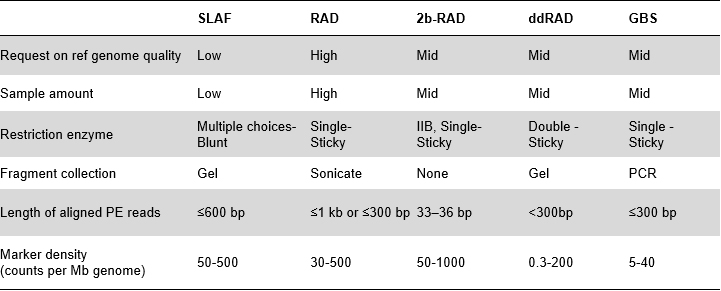
Výhody SLAF
Vyšší účinnost objevu genetických markerů– V kombinaci s vysoce výkonnou sekvenační technologií by SLAF-Seq mohl dosáhnout stovek tisíc značek objevených v celém genomu, aby splnil požadavky různých výzkumných projektů, ať už s referenčním genomem nebo bez něj.
Přizpůsobený a flexibilní experimentální design– Pro různé výzkumné cíle nebo druhy jsou k dispozici různé strategie enzymatického štěpení, včetně jednoenzymového, dvouenzymového a multienzymového štěpení.Strategie trávení bude předem vyhodnocena in silico, aby se zajistil optimální návrh enzymu.
Vysoká účinnost při enzymatickém trávení– Předem navržené enzymatické štěpení poskytuje rovnoměrnější distribuci SLAF na chromozomu.Účinnost sběru fragmentů může dosáhnout více než 95 %.
Vyhněte se opakované sekvenci– Procento opakující se sekvence v datech SLAF-Seq je sníženo na méně než 5 %, zejména u druhů s vysokou úrovní opakujících se prvků, jako je pšenice, kukuřice atd.
Samostatně vyvinutý bioinformatický pracovní postup– BMK vyvinula integrovaný bioinformatický pracovní postup použitelný pro technologii SLAF-Seq pro zajištění spolehlivosti a přesnosti konečného výstupu.
Aplikace SLAF
Mapa genetických vazeb
Konstrukce genetické mapy s vysokou hustotou a identifikace lokusů kontrolujících znaky květinového typu u chryzantémy (Chrysanthemum x morifolium Ramat.)
Časopis: Horticulture Research Publikováno: 2020.7

GWAS
Identifikace kandidátního genu spojeného s obsahem isofavonu v semenech sóji pomocí celogenomového mapování asociací a vazeb
Časopis: The Plant Journal Publikováno: 2020.08

Evoluční genetika
Populační genomická analýza a de novo sestavení odhalují původ plevelné rýže jako evoluční hry
Časopis: Molecular Plant Publikováno: 2019.5
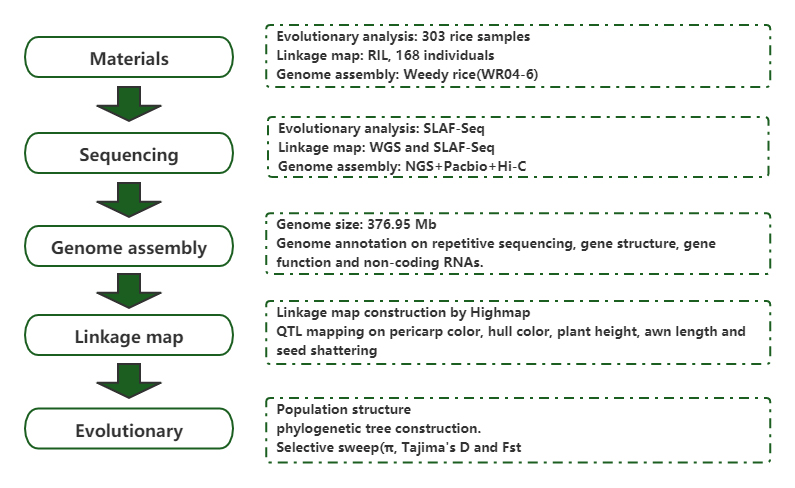
Hromadná segregační analýza (BSA)
GmST1, který kóduje sulfotransferázu, propůjčuje rezistenci vůči kmenům viru sójové mozaiky G2 a G3
Časopis: Plant, Cell&Environment Publikováno: 2021.04

Odkaz
Sun X, Liu D, Zhang X a kol.SLAF-Seq: účinná metoda rozsáhlého de novo objevování SNP a genotypizace pomocí vysoce výkonného sekvenování[J].Plos jedna, 2013, 8(3):e58700
Song X, Xu Y, Gao K a kol.Konstrukce genetické mapy s vysokou hustotou a identifikace lokusů kontrolujících znaky květinového typu u chryzantémy (Chrysanthemum × morifolium Ramat.).Hortic Res.2020; 7:108.
Wu D, Li D, Zhao X, a kol.Identifikace kandidátního genu spojeného s obsahem isoflavonů v semenech sóji pomocí celogenomového asociačního a vazebného mapování.Závod J. 2020;104(4): 950-963.
Sun J, Ma D, Tang L a kol.Populační genomická analýza a De Novo Assembly odhalují původ plevelné rýže jako evoluční hru.Mol Plant.2019;12(5):632-647.Mol Plant.2018;11(11):1360-1376.
Zhao X, Jing Y, Luo Z a kol.GmST1, který kóduje sulfotransferázu, propůjčuje rezistenci vůči kmenům G2 a G3 viru mozaiky sóji.Prostředí rostlinných buněk.2021;10.1111/ks 14066
Čas odeslání: leden-04-2022