

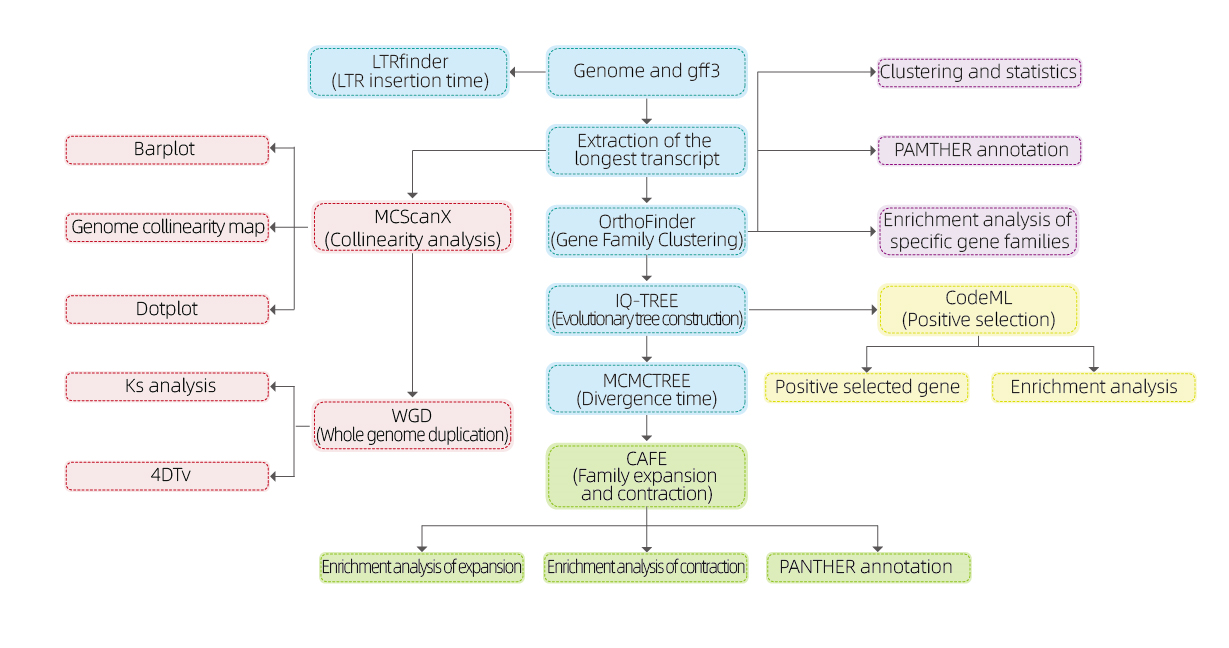
Comparative Genomics

Service Advantages

● Extensive Expertise and publication records: with accumulated, BMKGene has completed over 90 comparative genomics projects, with a cumulative impact factor reached 900.
● Comprehensive bioinformatics analysis: the analyses package contains the eight most commonly required analyses, providing well-designed ready-to-publish figures and allowing for a easy interpretation of the results
● Highly skilled bioinformatics team and short analysis cycle: with great experience in comparative genomics analysis, BMKGene’s team fulfills diverse personalized analysis demands in a short turn-around time
● Post-Sales Support: Our commitment extends beyond project completion with a 3-month after-sale service period. During this time, we offer project follow-up, troubleshooting assistance, and Q&A sessions to address any queries related to the results.
Service Specifications
|
Estimated turn-around time |
Number of species |
Analyses |
|
30 working days |
6 - 12 |
Gene family clustering Gene family expansion and contraction Phylogenetic tree construction Divergence time estimation(Fossil calibration required) LTR insertion time (For plants) Whole genome duplication (For plants) Selective pressure Synteny analysis |
Bioinformatics analyses
● Gene family
● Phylogenetics
● Divergence time
● Selective pressure
● Synteny analysis

Sample Requirements and Delivery
Sample Requirements:
Tissue or DNA for genome sequencing and assembly
For Tissue
|
Species |
Tissue |
Survey |
PacBio CCS |
|
Animal |
Visceral tissue |
0.5 ~ 1g |
≥ 3.5g |
|
Muscle tissue |
|||
|
≥ 5.0g |
|||
|
≥ 5.0mL |
|||
|
Mammalian blood |
|||
|
≥ 0.5mL |
|||
|
Poultry/Fish blood |
|||
|
Plant |
Fresh Leaf |
1 ~ 2g |
≥ 5.0g |
|
Petal/Stem |
1 ~ 2g |
≥ 10.0g |
|
|
Root/Seed |
1 ~ 2g |
≥ 20.0g |
|
|
Cells |
Cultured cell |
- |
≥ 1 x 108 |
Data
Genome sequence files(.fasta) and annotation files (.gff3) of closely related species
Service Work Flow

Experiment design

Sample delivery

Library construction

Sequencing

Data analysis

After-sale services
* Demo results shown here are all from genomes published with Biomarker Technologies
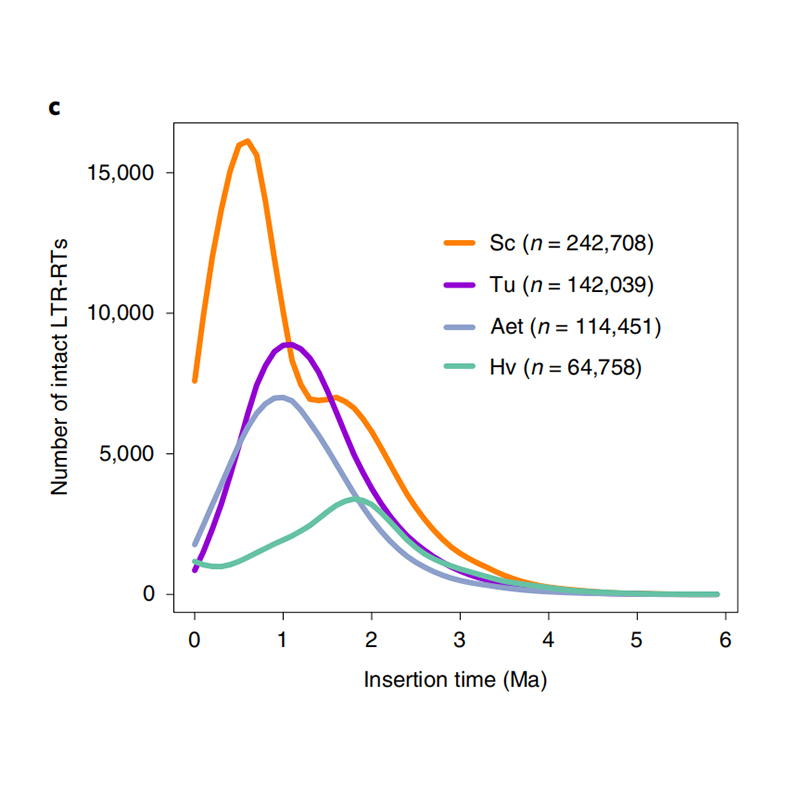
1.LTR insert time estimation: The figure shown a unique bimodal distribution in LTR-RTs insertion times in Weining rye genome, compared to other species. The most recent peak appeared around 0.5 million years ago.
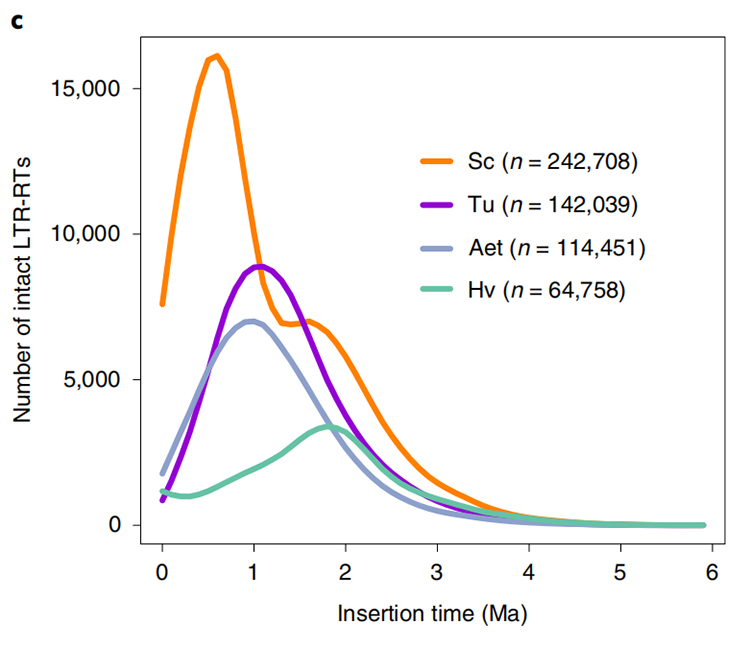
Li Guang et al., Nature Genetics, 2021
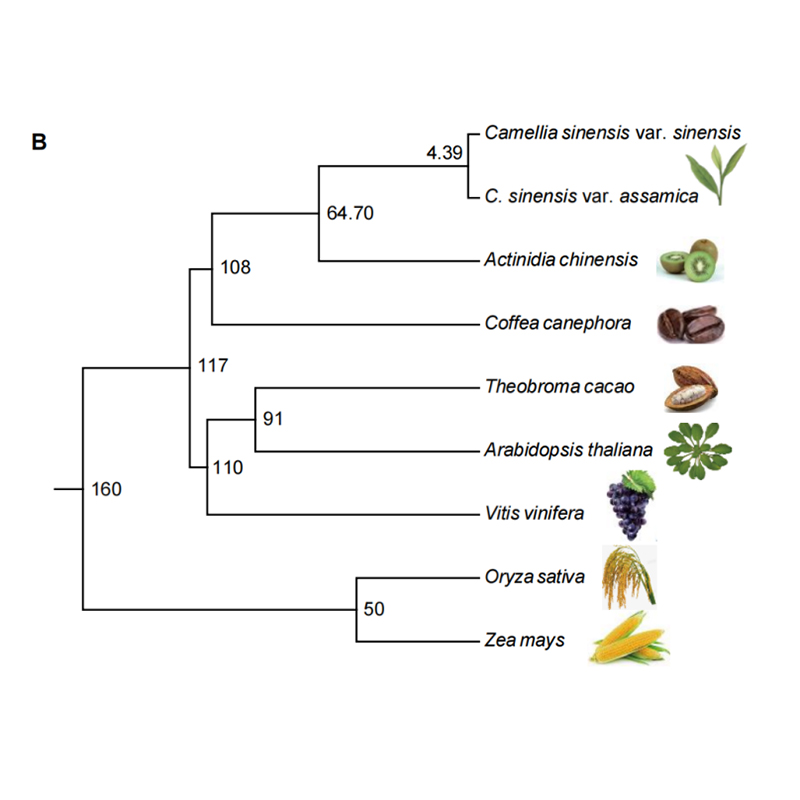
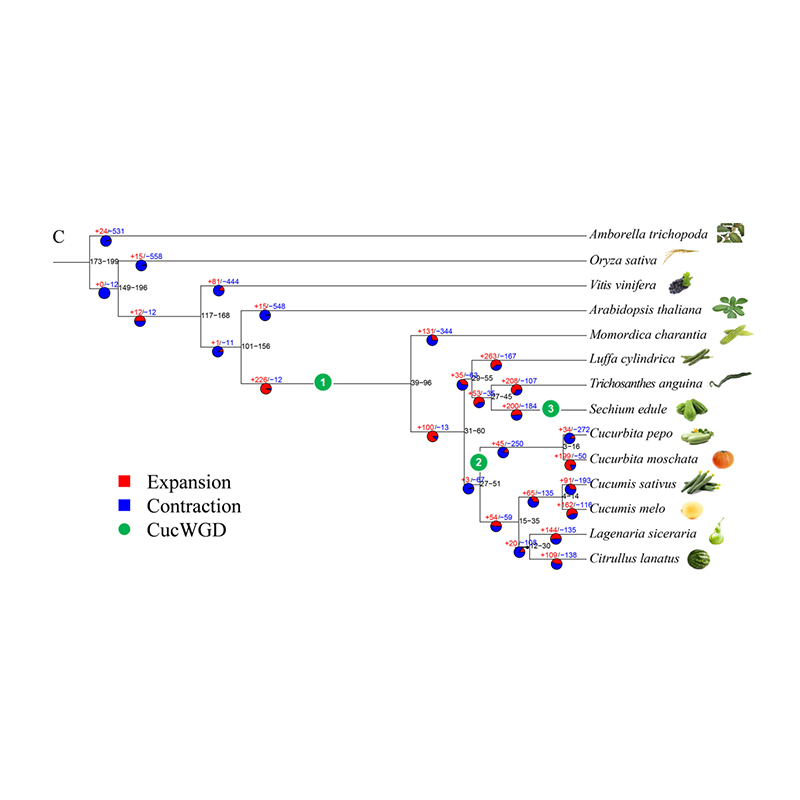
2.Phylogeny and gene family analysis on chayote (Sechium edule) : By analyzing chayote and the other 13 related species in gene family, Chayote was found to be most closely related with snake gourd (Trichosanthes anguina). Chayote derived from snake gourd in around 27-45 Mya and whole genome duplication(WGD) was observed in chayote in 25±4 Mya, which is the third WGD event in cucuibitaceae.
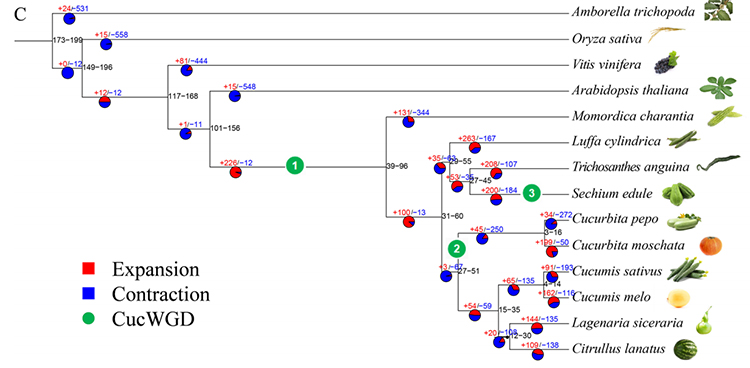
Fu A et al., Horticulture Research, 2021
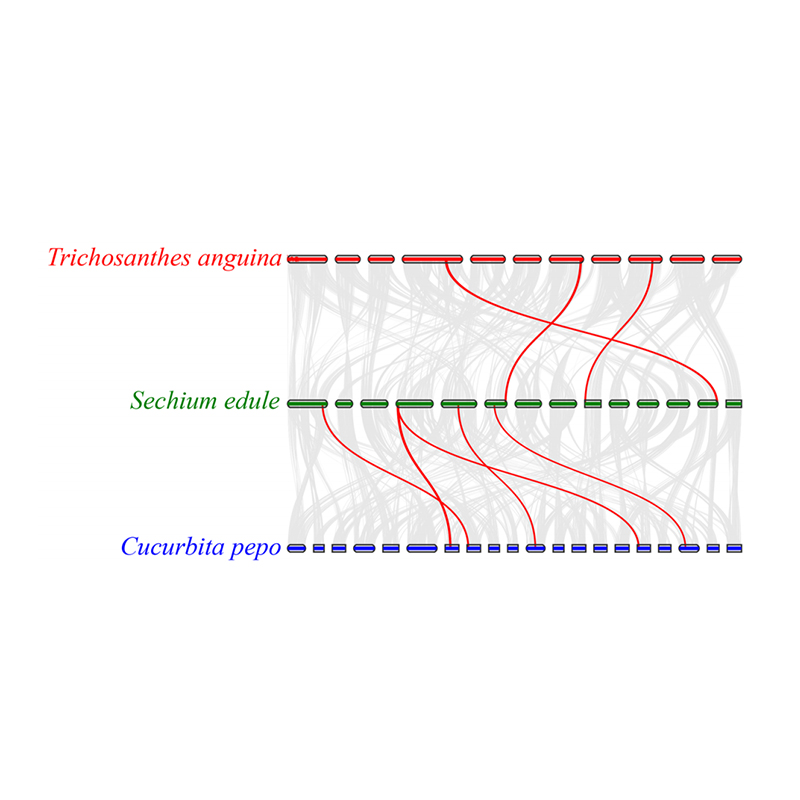
3.Synteny analysis: Some genes related to phytohormones in fruit development were found in chayote, snake gourd and squash. Correlation between chayote and squash is slightly higher than that between chayote and snake gourd.
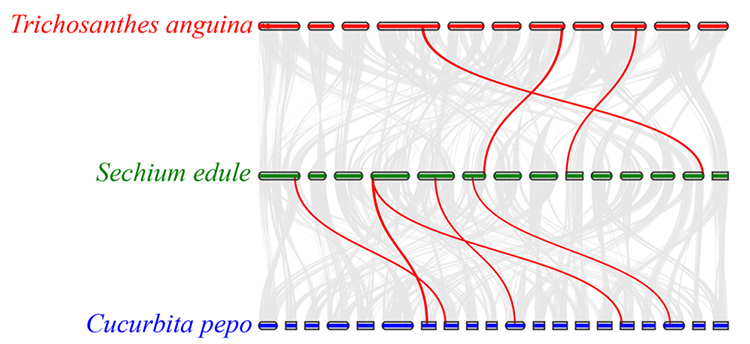
Fu A et al., Horticulture Research, 2021
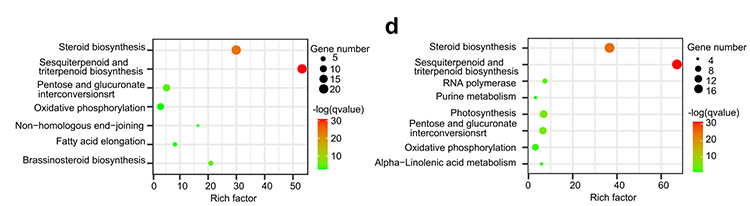
4.Gene family analysis: KEGG enrichment on gene family expansion and contraction in G.thurberi and G.davidsonii genomes shown that steroid biosynthesis and brassinosteroid biosynthesis related genes were expanded.
Yang Z et al., BMC Biology, 2021
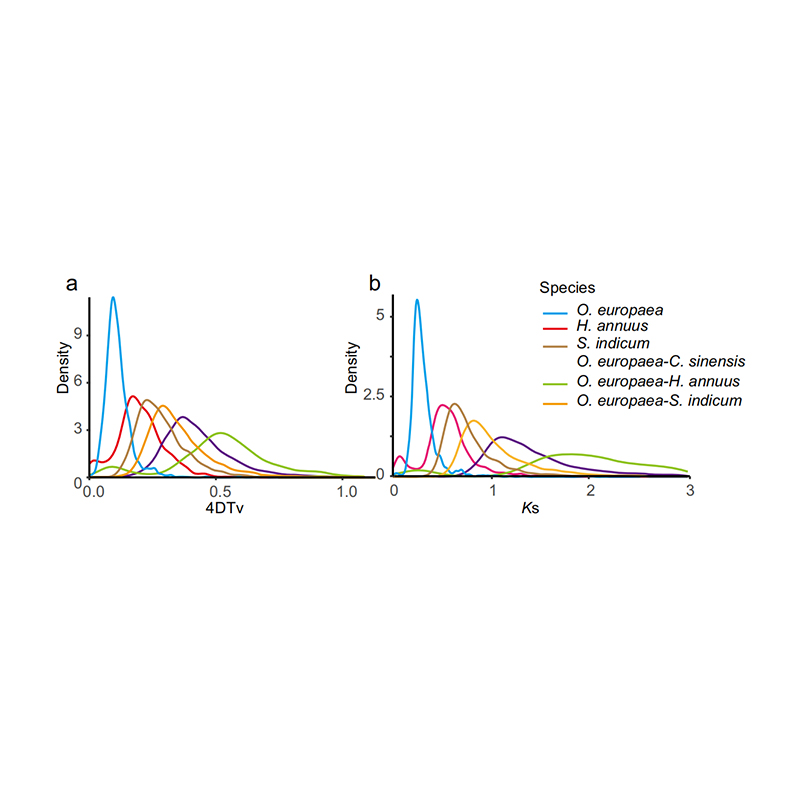
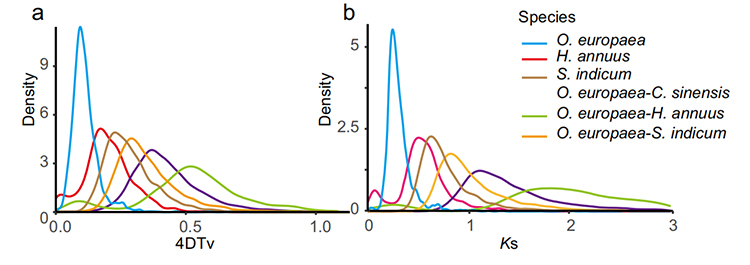
5.Whole genome duplication analysis: 4DTV and Ks distribution analysis shown whole genome duplication event. Peaks of intraspecies shown duplication events. Peaks of interspecies shown speciation events. The analysis indicated that comparing with the other three closely related species, O. europaea went through a large scale gene duplication more recently.
Rao G et al., Horticulture Research, 2021
BMK Case
Rose without prickle: genomic insights linked to moisture adaptation
Published: National Science Review, 2021
Sequencing strategy:
‘Basye’s Thornless’ (R. Wichurainan) genome:
Approx. 93 X PacBio + approx. 90 X Nanopore + 267 X Illumina
Key results
1.High quality R.wichuraiana genome was constructed using long-read sequencing techniques, which yield an assembly of 530.07 Mb (Estimated genome size was approximately 525.9 Mb by flow cytometry and 525.5 by genome survey; Heterozygosity was around 1.03%). BUSCO estimated score was 93.9%. Comparing with “Old blush” (haploOB), the quality and completeness of this genome was confirmed by base single-base accuracy and LTR assembly index (LAI=20.03). R.wichuraiana genome contains 32,674 protein coding genes.
2.Multi-omics joint analysis, consisting of comparative genomics, transcriptomics, QTL analysis of genetic population, revealed the crucial speciation between R. wichuraiana and Rosa chinensis. Also, expression variation of related genes in QTL were likely to be associated with stem prickle patterning.
Comparative genomics anaysis between Basye;s Thornless and Rosa chinensis including synteny analysis, gene family cluster, expansion and contraction analysis, revealed large number of variations, which related to crucial traits in roses. The unique expansion in NAC and FAR1/FRS gene family was very likely to be associated with resistance to black spot.
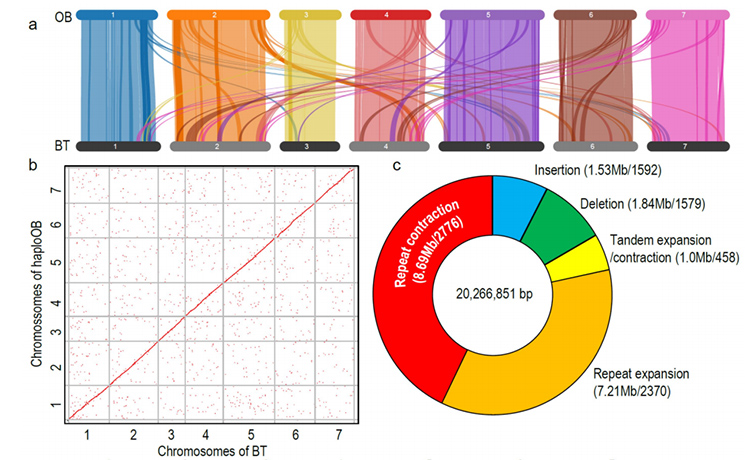

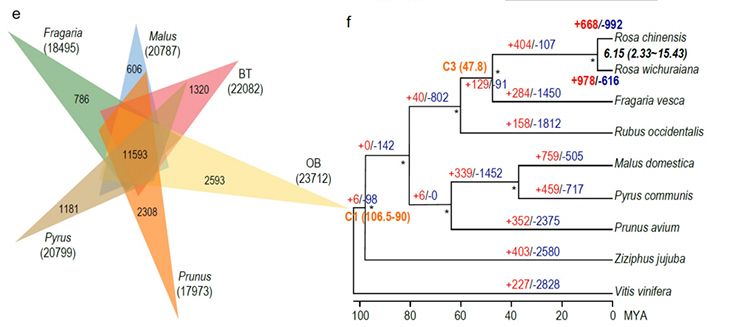
Comparative genomics analysis between BT and haploOB genomes.
Zhong, M. , et al. “Rose without prickle: genomic insights linked to moisture adaptation” National Science Review, 2021;, nwab092.





